Pre-IND:
依据《国家药品监督管理局关于调整药物临床试验审评审批程序的公告》:申请人在提出新药首次药物临床试验申请之前,应向药审中心提出沟通交流会议申请。其中,在申请人递交IND申请前,可自愿进行一次重要的会议,即Pre-IND会议。
可在临床研发不同阶段提出沟通交流申请,沟通交流会分为I类、II类、III类三种会议类型,每类会议针对不同情况开展,Pre-IND会议属于II类会议。从申请人申请到召开会议,I类会议需要在30日内,II类会议需60日内,III类会议需75日内开展。
二、NDA
NDA(New Drug Application)是指新药经过临床试验后,申报注册上市的阶段;主要目的是确保上市药品安全有效和质量可控,经过NDA,药品获得批准后才能上市销售。
根据《药品注册管理办法》:“申请人在完成支持药品上市注册的药学、药理毒理学和药物临床试验等研究,确定质量标准,完成商业规模生产工艺验证,并做好接受药品注册核查检验的准备后,提出药品上市许可申请。”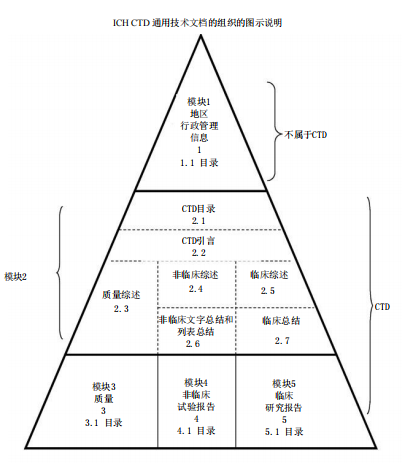
一般要求按照通用技术文件(CTD,Common Technical Document)的格式来组织和提交文件。
FDA关于NDA内容,可查看:https://www.fda.gov/drugs/types-applications/new-drug-application-nda。
Pre-NDA:
Pre-NDA会议(新药上市申请前会议)是药品上市许可申请前的重要沟通交流会议,申请人在进行Pre-NDA会议沟通交流时,需明确会议目的、提出具体的沟通交流问题、充分准备资料和研究数据,以解决NDA申报前存在的关键技术问题。
BLA:
BLA(Biologic License Application)生物制品许可申请:大部分的生物医药制品是采取BLA途径,NDA主要适用于化学药品。
以上是新药(创新药)产生必须进行的两项行政审批,不仅创新药上市需要审批,仿制药的上市批准也需要审批。